ALL: Zdravim vsechny. Mam tu zase dotaz, ale uvedomuji si ze je lepsi ho dat sem nez do auditka
[ Chemia ] .
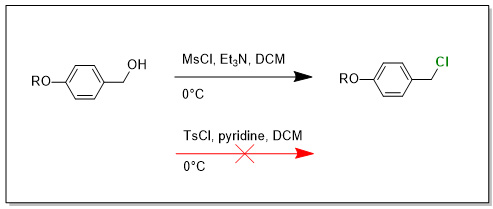
Mam tu problem s jednoduchou mesylaci, tosylaci.
Zkousel jsem ruzne podminky, treba - 20°C pro MsCl, nebo +40°C pro TsCl. Tam uz (TsCl/40°C) trochu RO-Ph-CH2Cl vznika (ale ten mi nepotrebujeme pac chlorid neni reaktivni v dalsich krocich), ale pri nule ci r.t. prakticky jen vychozi latka, obcas stopy produktu. Nejaky napad na co se zamerit pri optimalizaci? Sazmozrejmne kdyz vezmu vsechny komponenty co mam, stejne usporadani experimentu, stejne lahvicky tak s libovolnym benzylovym alkoholem ze skrine mam 100% konverzi na mesylat a 90+% preparativni vytezek. Jen s timhle konkretnim zatracenym alkoholem mam problem. R je elektronove bohaty benzyl aromat. Tzn.: ArCH2-O-Ph-CH2-OH
Prijde mi ze v pripade mesylace bude mechanismus nekde kolem reakce RO-Ph-CH2-OMs s Et3N za vzniku kvarterni ammoniove soli a nasledny rozklad za ucasti chloridoveho aniontu. Nebo tak neco.
Ale Et3N se dava 1.2 - 1.5 ekvivalentu. To by znamenalo ze to reaguje az s Et3N.HCl, protoze je tam 80+% konverze na ten RO-Ph-CH2Cl.
Achjo, tohle mel student na 5 tydenni Internship a tohle byl posledni step ze 6 kroke syntezy co se mu tak hezky darila :-( Vypadam pak jak strasnej reznik co dava studentum jen speky na kterych si vylamou zuby.